NTRK Inhibitors
New Options for Tissue-Agnostic Treatment
NTRK inhibitors work to target tyrosine kinase genes, rather than killing cells based on morphology or body site. NTRK drugs can be grouped into multi-kinase inhibitors, which are active against a range of kinase-encoding genes, or more selective NTRK-specific inhibitors. Multikinase inhibitors include entrectinib, crizotinib, cabozantinib, lestaurtinib, altiratinib, foretinib, ponatinib, nintedanib, merestinib, MGCD516, PLX7486, DS-6051b and TSR-011, while the only currently FDA-approved NTRK-specific inhibitor is Larotrectinib.
Entrectinib
Of the multikinase inhibitors, entrectinib has demonstrated the most promising response in clinical trials. The drug has been tested in several clinical trials so far:
Study:
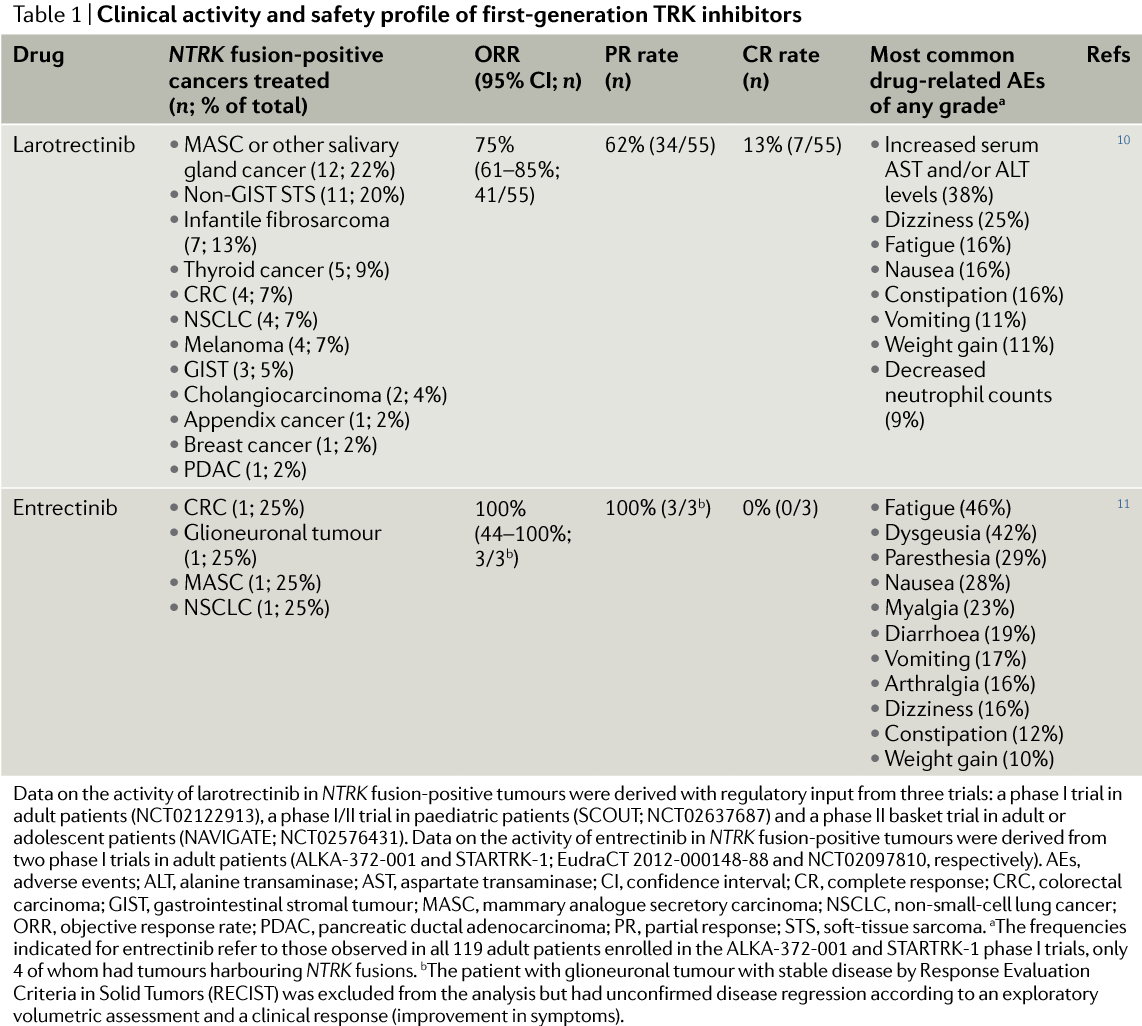
A set of combined phase I studies on adults with: (1) NTRK, ROS1 or ALK rearrangements and (2) NTRK, ROS1, and ALK abnormalities (point mutations, amplifications, copy-number variants or insertions/deletions). In these two studies, 4 patients with NTRK fusion-positive tumors were treated with entrectinib, one with colorectal carcinoma, one with MASC, one with lung adenocarcinoma and one with glioneural tumour.
Results:
The first three patients achieved partial responses, and the patient with glioneuronal tumor had substantial disease regression. Overall, the ORR in TKI treatment-naive patients with cancers harbouring an NTRK, ROS1 or ALK fusion were 100% (3 of 3 patients), 86% (12 of 14 patients) and 57% (4 of 7 patients), respectively.
Study:
A phase I/Ib study (STARTRK-NG, NCT0265040) in children and young adults (aged 2–22 years) with or without NTRK, ROS1 or ALK fusions.
Results:
Currently recruiting, results not yet published.
Study:
A phase II basket trial (STARTRK-2, NCT02568267) in adults with NTRK, ROS1 or ALK fusions.
Results:
Currently recruiting, results not yet published.
Larotrectinib
The activity of the TRK-selective inhibitor larotrectinib in patients with tumours harbouring NTRK fusions has been explored in three clinical trials:
- A phase I trial in adults (NCT02122913)
- A phase I/II trial in pediatric patients (SCOUT, NCT02637687)
- A phase II trial involving adults and adolescents (NAVIGATE, NCT02576431)
Combined results from first 55 evaluable patients have been compiled. Patients with a total of 17 unique cancer types harbouring NTRK fusions, including MASC, infantile fibrosarcoma, melanoma, GIST, and thyroid, colorectal or lung adenocarcinoma, were included in this analysis. :
- The overall response rate (ORR) was 75% and the complete response rate (CRR) was 13%.
- The median time to response was 1.8 months.
- Responses were observed agnostic of tumor tissue, fusion type (NTRK1, NTRK2 or NTRK3), upstream partner, and age (patients from 4 months to 76 years were treated).
- Median progression-free survival duration had not yet been reached, but at the last data cut-off, 55% of the patients were still progression-free after 1 year of treatment, and 71% of responses were ongoing.
Published in Nature Reviews Clinical Oncology 2018
NTRK fusion-positive cancers and TRK inhibitor therapy
Emiliano Cocco, Maurizio Scaltriti, Alexander Drilon
Mechanisms of Resistance
- Because TRK inhibitors are relatively new treatment options, there’s limited data on acquired clinical resistance to these drugs.
- Currently, point mutations in the fusion’s NTRK kinase domain is the only mode of secondary resistance detected in TKI-resistant patients, although it’s likely that off-target or bypass resistance mechanisms also exist.
- Resistance mutations can result in amino acid substitutions involving the solvent front, activation loop xDFG motif or so-called gatekeeper residue of TRKA or TRKC. No TRKB mutations have yet been detected, likely due to the limited number of patients with NTRK2 fusions evaluated.
- The first case of acquired resistance to TRK inhibition was reported in 2016 in a patient with LMNA–NTRK1 fused colon cancer after 4 months of treatment with entrectinib. Resistance was found to be caused by two mutations resulting in substitutions in the kinase domain of TRKA — G595R and G667C.
References:
•
Cocco E, et al. (2018) Nat Rev Clin Onc: 1.